Description
fastkmers is a simple program for getting k-mer counts from a fastq/fasta file, written in Rust.
It takes a fastq/fasta file as input and outputs the counts of k-mers of a specified length. It is implemented using hash tables and a simple algortihm but is still reasonably fast. The maximum supported k-mer size is 31.
Install
I provide precompiled binaries for linux only here, but it is simple to compile and run:
curl --proto '=https' --tlsv1.2 -sSf https://sh.rustup.rs | sh
git clone https://github.com/angelovangel/fastkmers.git
cd fastkmers
cargo build --release
The executable file fastkmers is now under ./target/release/
Usage
# Make sure the executable is in your path
# check available options
fastkmers -h
# to get 4-mer counts and a summary
fastkmers -k 4 -s file.fastq.gz
# output json, input fasta
fastkmers -k 4 -a -j file.fasta
# query for a specific k-mer
fastkmers -k 5 -q "AATTG" file.fastq.gz
# query with regex is also supported
# this example would match k-mers whose last 4 bases are: not T| A | T or G | A
fastkmers -k 5 -q "[^T]A[T|G]A$" file.fastq.gz
The k-mer counts are printed to stdout as a tab-separated table or as json.
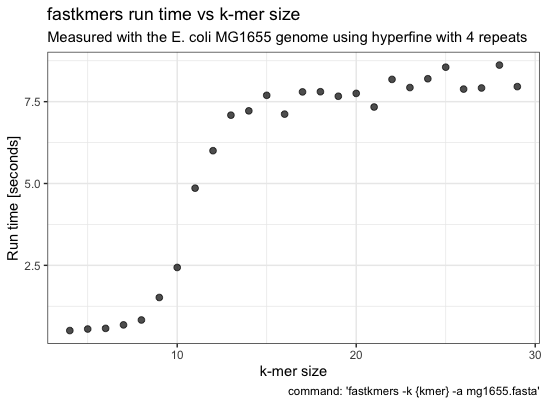
Speed
I haven’t compared to other programs (e.g. jellyfish), below are some measurements of the execution times for different k-mer sizes of the E. coli MG1655 genome, performed on a MacBook Pro 2018 (Intel i5 and 8 Gb RAM).
hyperfine -r 4 --warmup 1 --export-csv hyperfine-kmer-size.csv -P kmer 4 29 'fastkmers -k {kmer} -a mg1655.fasta'